No time to read now?
-> Download the article as a handy pdf
List of contents
What are Periodic Safety Reports?
Understanding Aggregate Reporting in Pharmacovigilance
Martti Ahtola | Oct 16, 2023

It can seem that there are several acronyms related to periodic safety reporting, that each country and region has different requirements and that in general, aggregate reporting is a big hassle. We want to clarify and demystify this process as much as possible. We start from the very basics and go all the way into the nitty-gritty details.
- Periodic safety reports, or aggregate reports, are one of the backbones of pharmacovigilance activities.
- Periodic safety reporting starts from the beginning of the first clinical phase of the drug development process and continues throughout the rest of the phases of the clinical trials.
- The final results of the collection of safety reports from the clinical trials are evaluated when the documentation for marketing authorization application is submitted to the regulators.
- Periodic safety reporting continues once the application is approved and the responsibility to regularly assess and report safety information lasts as long as the authorization is valid.
Because the purpose of this text is to give the reader a comprehensive understanding of periodic safety reporting of medicinal products while being suitable for both newcomers and pharmacovigilance experts, there can be different ways to go through the text.
The first section describes the basic principles of pharmacovigilance and what periodic reporting means in this context, so if you know, or think you know, your stuff when it comes to the basics of drug safety activities, feel free to move directly to the next part of the text or jump to the part you are interested in by using the table of contents.
Pharmacovigilance and Periodic Safety Reports
The book description of pharmacovigilance is:
“Science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other medicine-related problem.”
Our layman’s description could be for example:
“Making sure your drug is safe to use.”
Or
“Ensuring that the benefits of the medicine outweigh the risks.”
Another way to explain what pharmacovigilance is, could be a list of the activities that are involved. This list could include for example these activities:
- Monitoring the safety of the medicinal products
- Collecting adverse event reports from patients and healthcare professionals
- Reporting safety issues to the authorities
- Regularly assessing the safety-benefit profile of the drug
- Identifying and mitigating the risks related to the product
- Reviewing literature for the latest safety-benefit information
All of these activities are tied to periodic safety reports in one way or another, because essentially periodic safety reports are a summary of the product’s status related to the patient population, performed activities, new information learned and the current understanding of the risks and benefits.
What is Drug Safety Reporting?
Safety reporting consists of collection of adverse event reports and reporting of adverse reactions of drugs to the regulatory authorities.
An often used term in safety reporting is Individual Case Safety Report (ICSR) which refers to a single report of an adverse event related to one or more drugs, and one patient. Terms such as adverse event report, adverse drug reaction report, safety report, and other similar report names are usually used as synonyms for ICSR.
Pharmaceutical companies and regulatory agencies must ensure they have a good set of processes, a “system”, in place to capture all suspected adverse reactions, store them in a safety database, validate as ICSRs, and share with regulatory authorities and/or other pharma companies. This is known as pharmacovigilance system or PV system.
The term “PV system” should not be confused with PV software, such as safety databases or pharmacovigilance platforms (computerized systems).
In general, there are two main pathways for safety reporting:
1. SUSARs/ICSRs
2. Aggregate Reports
This text is mainly about the second pathway, aggregate reports, but it is important to understand that the aggregate reporting consists of a collection of ICSRs and the analysis of the aggregate data.
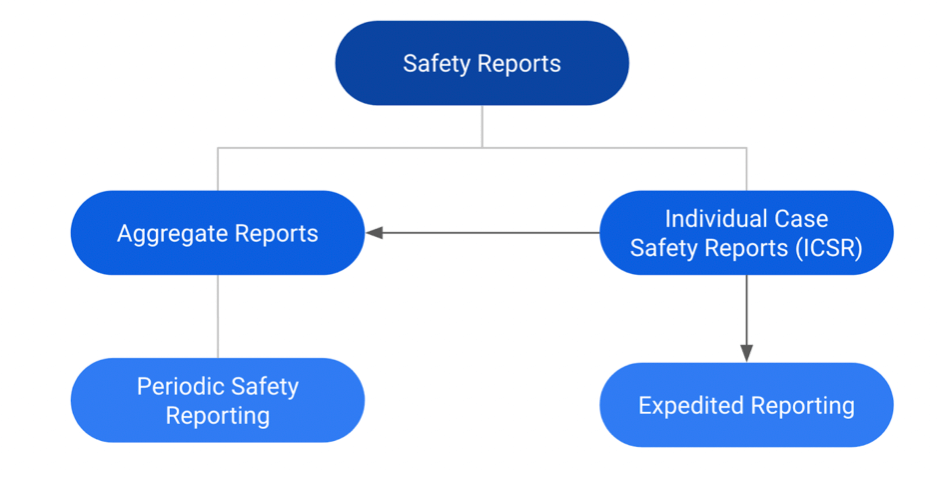
Diagram showing the basic structure of safety reports
What are the sources of Safety Information?
Both marketing authorization holders and regulatory authorities have a responsibility to collect and report forward adverse event reports. So where do the individual adverse event reports come from?
Unsolicited Reports
Unsolicited reports can be received in theory from anywhere, for example from:
- Patients communicating via medical enquiries (website, phone…)
- Company representatives
- Healthcare professionals
- Social media (and anywhere on internet)
- Scientific literature
- And many other additional sources.
Solicited Reports
Solicited reports are typically from organized data collection programs, such as:
- Clinical trials
- Non-interventional studies
- Early access programs
- Compassionate use
- Patient support programs
- Surveys
- And other organized data collection systems.
If pharmacovigilance and drug safety is a new topic for you but you are determined to get a full understanding of the aggregate safety reports, in addition to this quick intro, you can also read our introduction to pharmacovigilance first before proceeding further in this text.
Timelines for Safety Reporting
The first thought related to adverse event reporting is often tight timelines such as 24 hours or 7/15/30/90 days (depending on seriousness and other factors).
Basically, this is the case with all ICSRs, but in addition to this “expedited reporting”, most safety reports are also collected to a safety database and then further analyzed in Signal Detection and Periodic Safety Reports.
The development of the understanding of a medicinal product’s safety profile is monitored and evaluated by the sponsor of the clinical trial or license / marketing authorization holder by looking at the continuously growing body of safety related information from the individual reports in different analytical ways.
Results of this evaluation are then put in a periodic aggregate report and then submitted for a review by the regulatory authorities.
What kinds of periodic safety reports are there?
The main periodic reports related to drug safety are DSUR, PBRER/PSUR/PADER and signal detection reports*. Here we will concentrate on DSUR and PBRER (and their variants). We’ll go into detailed descriptions of these reports and look at what the requirements are, what the reports contain, how they are prepared and when and how they should be submitted.
*For signal detection, an internal report is often produced, but this report is not submitted to the regulatory authorities. Only in situations where a marketing authorization holder detects and validates a signal for new safety-related information about their product (that has not been detected by the regulatory authorities or other marketing authorization holders), they inform the regulatory authorities but this happens usually with a simple form and/or through an email communication, instead of submitting the internal signal detection report.
Development Safety Update Report (DSUR)
The idea of the DSUR is for it to be broadly useful, including not only the safety-related information but also an overview into the drug development and investigation. The DSUR format is the result of the work of the international council of harmonization (ICH). The DSUR guideline that is part of the Efficacy and Pharmacovigilance category, was published in 2010.
The ICH guideline defines the recommended content and format of a DSUR and provides an outline of points to be considered in the preparation and submission of the report.
The harmonization of the content, format, and timing of periodic safety reports should help to ensure that regulatory authorities in the ICH regions receive a uniform, high-quality, comprehensive report. ICH has published the E2F Development safety update report scientific guideline that is followed at least in Canada, China, Europe, Japan, Singapore, Switzerland, Turkey, the United Kingdom and the United States (= the ICH regions).
Before implementation of the DSUR format, the regulation of some ICH countries and regions required submission of periodic safety reports to regulatory authorities to provide this information. The challenge was that there were significant differences in the content, format and timing of these reports. Some countries and regions also required a periodic report describing the status of ongoing investigations, manufacturing changes, and overall development status and plans.
Purpose of the DSUR
The main objective of a DSUR is to present a comprehensive, thoughtful annual review and evaluation of pertinent safety information collected during the reporting period related to a drug under investigation by:
- Examining whether the information obtained by the Sponsor during the reporting period is in accord with previous knowledge of the investigational drug’s safety
- Describing new safety issues that could have an impact on the protection of clinical trial subjects
- Summarizing the current understanding and management of identified and potential risks
- Providing an update on the status of the clinical investigation/development program and study results.
Format and Frequency of DSURs
As described above, US FDA, EMA in the EU and PMDA in Japan and several other main authorities in the largest pharmaceutical markets consider the Development Safety Update Report (DSUR) to be a common standard for periodic reporting on products under development (including marketed drugs that are under further study). DSUR should meet national and regional requirements in the ICH regions replacing any local safety reports that have been used in those markets before implementation of DSUR.
The ICH E2F guideline provides instructions on the format and contents of the report. The end result is a pdf document with a text section and annexes with aggregate data tables.
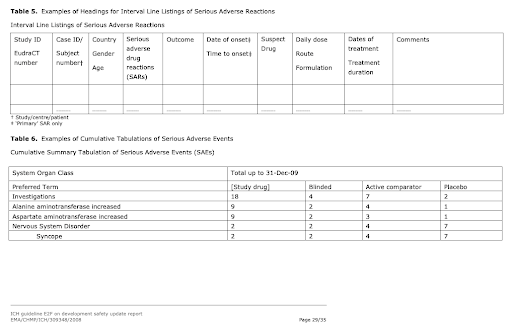
Aggregate data tables in the DSUR that are produced from the Safety Database
DSUR is submitted annually. The exact submission date depends on the data lockpoint date of the DSUR, which is defined by the Development International Birth Date (DIBD). The DSUR should be submitted to all concerned regulatory authorities no later than 60 calendar days after the DSUR data lock point.
Aggregate data tables can be easily generated with Tepsivo Safety Database
Periodic Safety Reporting in Postmarketing Phase
Once the marketing authorization has been granted for the product, the periodic safety reporting requirements continue but the report type(s) changes. It is possible that the DSUR reporting continues simultaneously with PSUR reporting if there is an obligation to conduct a post-authorization safety study (PASS), or there are additional clinical trials for different indications, formulations etc.
Periodic safety reporting responsibilities in the post-marketing phase can be seen as a continuation of the reporting activities during clinical studies. However, there are major differences, mainly because the amount of new data per period can increase, data sources are more varied, the exposure can increase significantly, the quality of the data in general decreases, the amount of data received per patient is significantly lower, and the duration of this responsibility is indefinite.
Harmonization of report format and reporting frequency is in place in the post-marketing phase as well but as during the clinical phase, there might be differences between the regions and countries. Similarly, as with the DSUR, the big pharmaceutical markets (ICH members) mostly follow the common guideline for PBRER but there can be country-specific differences in schedules and formats.
PBRER, PSUR and PADER
What is a Periodic Benefit-Risk Evaluation Report (PBRER)?
The PBRER is intended to be a common standard for periodic benefit-risk evaluation reporting on marketed products, including approved drugs that are under further study, among the ICH regions.
Periodic benefit-risk evaluation report is a pharmacovigilance document intended to provide an evaluation of the risk-benefit balance of a medicinal product at defined time-points after its authorization.
PBRER’s objective is to present a comprehensive and critical analysis of the risk-benefit balance of the product taking into account new or emerging safety information in the context of cumulative information on risk and benefits.
The ICH E2C(R2) Guideline for Periodic Benefit-Risk Evaluation Report is followed by Brazil, Canada, China, Europe, Japan, Korea, Mexico, Saudi Arabia, Singapore, Switzerland, Turkey, UK, and the United States.
What is a Periodic Safety Update Report (PSUR)?
Periodic Safety Update Report (PSUR) is a pharmacovigilance document intended to provide an evaluation of the risk-benefit balance of a medicinal product at defined time points after its authorization. Objective of the PSUR is to present a comprehensive and critical analysis of the risk-benefit balance of the product, taking into account new or emerging safety information in the context of cumulative information on risk and benefits.
Sounds familiar? We’ll get to that soon.
With the recognition that assessment of the risk of a medicinal product is more meaningful when evaluated in context of its benefits, the guidelines were revised with change in the report format from PSUR to PBRER in 2012.
What is a Periodic Adverse Drug Experience Report (PADER)?
Periodic Adverse Drug Experience Report (PADER) is a type of aggregate safety report required to be submitted by a sponsor or marketing authorization holder (MAH) to the US Food and Drug Administration (FDA) after obtaining marketing authorization approval.
It means that the requirement to submit a PADER starts following completion of clinical studies and with approval of a new drug application (NDA for innovator products), abbreviated NDA (ANDA for generic products), and biologic license application (BLA for biological products) by the US FDA.
For each NDA, ANDA, and BLA, MAH should submit a separate PADER. PADER is required to be submitted by a Marketing Authorization Holder (MAH) to the FDA after U.S. commercialization.
Although as per 21 Code of Federal Regulations (CFR) 314.80 US FDA recommends periodic submission of a PADER, the MAH can submit PSUR/PBRER along with NDA listings (also called US Supplement/FDA PSUR) in place of PADER after obtaining a waiver per 314.90 and 600.90.
FDA accepts all three formats, the PADER/PAER, PSUR, and PBRER, to fulfill the postmarketing periodic safety reporting requirements under 314.80(c)(2) and 600.80(c)(2). Each format must be submitted according to the content and timelines specified in the US regulations (PADER/PAER) or by ICH (PSUR and PBRER).
What is the difference between PADER and PAER?
PAER stands for periodic adverse experience report. PAER is mentioned together with PADER in the FDA documents covering periodic reporting. Often PADER and PAER are mentioned in the FDA documents in a way that seems to suggest that they are interchangeable. As an example, the section above that has been summarized from FDA website and documents where PADER is always written in the format “PADER/PAER”. The reference to the format of PAER is the same part of 21 CFR 600.80(c)(2) as for PADER.
Which one to submit: PBRER, PSUR or PADER?
The rule of thumb for periodic safety report submission is the following: PADER for the US and PBRER (PSUR) for the rest of the world.
As mentioned above in the PSUR description, PSUR was actually replaced with the PBRER in 2012. Then why do we even talk about PSUR?
“PSUR” as a term is still commonly used in the EU but at least format-wise it actually means PBRER. The PBRER format replaced the previously used PSUR format but to align the terminology with the EU legislation and perhaps to continue with the established terminology, the report is described as PSUR in the GVP Modules. It is important to remember that this EU PSUR is in fact a PBRER.
FDA accepts all three formats, the PADER, PSUR, and PBRER, to fulfill the postmarketing periodic safety reporting requirements. Each format must be submitted according to the content and timelines specified in the regulations or by ICH.
The ICH E2C(R2) guidance introduced new concepts linked to an evolution of the traditional Periodic Safety Update Report (PSUR) from an interval safety report to a cumulative benefit-risk report.
PBRER provides greater emphasis on benefit than the previously used PSUR, particularly when risk estimates change importantly. In such cases, there will need to be an overall explicit evaluation of benefit-risk. This is emphasized by the name of the report, “Periodic Benefit-Risk Evaluation Report” (PBRER). The PBRER template also provides greater emphasis on the cumulative knowledge regarding a medicinal product than the previous PSUR format. PBRER still retains the focus on new information that has been collected during the previous interval.
A formal evaluation of benefit was a new feature of the PBRER. In practice a concise discussion of benefit will usually be sufficient, unless the safety or benefit-risk profile has changed significantly during the reporting interval.
Requirements for periodic safety reporting
during the life-cycle of a drug
Periodic safety reporting is required throughout the life-cycle of the medicinal product. The requirements start when the first clinical trial is approved and end when the last license expires.
Below, we offer a summary of the periodic safety report requirements from the lifecycle perspective, grouping the lifecycle into:
- Clinical Trials
- Marketing Authorization Application
- Postmarketing
Clinical trials
DSUR – ICH E2F
Periodic reporting is part of clinical trials in different phases and end points, starting from the safety phase I. During clinical trials, there are legal requirements for annual safety reporting for the Sponsor. Annual Safety Reports are sent to the regulatory bodies by the sponsor based on the legal requirements of the country and guidelines from the responsible regulatory body.
The main aggregate report for safety during clinical studies is the Development Safety Update Report (DSUR). Often the DSUR and the annual safety report are one and the same.
Post-approval
PADER / PBRER / PSUR – ICH E2C(R2)
Pharmacovigilance activities, including periodic reporting, are required as long as there is an effective authorization irrespective of the marketing status of the product.
Depending on the market it is normal that after an originator product (not generic, biosimilar etc.) has been approved, an annual aggregate safety report, PSUR, is required.
How to prepare a periodic safety report?
Content and Format
Templates and guidance on the content available for DSUR, RMP and PADER / PSUR / PBRER. Usually, there is a Word template available and/or the required sections are described in the applicable legislation and guidelines.
To put it simply, the finalized periodic safety report is a signed pdf text document.
DSUR Format
DSUR is a common standard for periodic reporting among the ICH regions but the DSUR should meet the national and regional requirements in the ICH regions replacing any local safety reports that have been used in those markets before implementation of DSUR.
DSUR format is not defined in the EU or US legislation the same way as the PBRER/PSUR content (see below). The EU regulation 536/2014 on clinical trials (Article 43) on medicinal products for human use describes that the sponsor is required to submit annually through CTIS a report on the safety of each investigational medicinal product used in a clinical trial but there are no specific details in the regulation. The regulation does say that the ICH members should follow the practices agreed by the council, meaning that DSUR template should be used.
The EU regulation on clinical trials also states that the annual report shall only contain aggregate and anonymized data. The regulation also highlights that the annual reports shall contain Reference Safety Information (RSI) that is in effect in the beginning of the reporting period and any significant changes to the RSI shall be described in the report and revised RSI shall be another annex in the annual safety report.
US 21 CFR 312.33 on Annual reports states that a sponsor shall submit annually a summary of all IND safety reports submitted during the past year. There is a proposed update (Docket FDA-2020-N-0258) to this rule from the FDA where the current annual reporting requirement would be replaced with annual DSUR.
In the box below, there is an ICH example table of contents for commercial sponsor’s DSUR.
PBRER format
PBRER has been developed in such a way that the content of several sections can be used for sections of other documents as a basis for a modular approach. For example, if the Development International Birth Date (DIBD) of a DSUR for a medicinal product is aligned to the international birthdate (IBD) of the PBRER for the same product as suggested in ICH E2F, the content of a number of sections of the DSUR can also be used in the PBRER when the data lock points (DLPs) are the same (i.e., when each report covers an interval of 1 year based on the IBD).
In the EU, the Article 35 of the Commission Implementing Regulation describes the structure of PSUR and Module VII of the Guidelines on Good Pharmacovigilance Practices (GVP) provides guidance on the preparation, submission and assessment of PSURs.
The EU Directive defines that PSUR contains:
Summaries of data relevant to the benefits and risks of the medicinal product, including results of all studies with a consideration of their potential impact on the marketing authorisation.
A scientific evaluation of the risk-benefit balance of the medicinal product. The evaluation must be based on all available data, including data from clinical trials in unauthorized indications and populations.
All data related to the volume of sales of the product and any data the MAH has related to the volume of prescriptions, including an estimate of the population exposed to the product.
PBRER/PSUR structure in the European legislation
Following format is a legal requirement presented in Implementing Regulation 520/2012 Annex II for both nationally authorized products and centrally authorized products. Click the box below to expand the full structure.
Additional information required
In addition to above described format and content, the PBRER dossier should contain a cover letter, including the formatted table template.
Dates and Timelines
For periodic safety reporting, the time period is essential. There are quite a few different dates that are relevant:
- Development International birth date
- International birth date
- Data lockpoint
- Submission due date
- Frequency of reporting
- Assessment date
Development International Birth Date (DIBD)
Development International birth date is the date when the first clinical study related to the investigational medicinal product has been approved to start by a regulatory authority anywhere in the world.
International birthdate (IBD)
International birth date is the date of the first approval of marketing authorization. IBD can be synchronized with DIBD which makes it easier to continue the periodic reporting and reduces the workload if both DSUR and PSUR are required to be prepared.
Data lockpoint (DLP)
Data lock point is the last day of the time period for the report. This means that only data generated before or on this date is used for the report.
Submission due date
There is a timeline to prepare and submit the periodic report after the data lockpoint. This timeline is usually 60 or 90 days. In the US, the sponsor is responsible for submitting the annual reports within 60 days.
Frequency of reporting
The frequency is not exactly a date, but the frequency affects the DLP and submission due date.
Read more about the frequency of reporting in the dedicated chapter below.
Assessment dates
For example in the European Union for centrally approved products, the Good Pharmacovigilance Practice module VII defines that the Preliminary Assessment Report (PRAR) should be made available in 60 days from submission for centrally approved products, there are 30 days for the MAH(s) and PRAC to comment the PRAR, 15 days for the Rapporteur to deliver an updated report based on the comments etc.
Previous Reports
One of the key things that affect the process of preparing a periodic safety report is whether it is the first report or not. In practice, this means the first DSUR because as mentioned above, information from the DSUR can be reused in the PBRER preparation. So if there are previous DSURs and/or PBRERs available, the preparation of the periodic report should be an update of the previous version of the report. The transition from DSUR to PBRER is not as simple as just updating the information to a new template. While information from the latest DSUR can and should be utilized, preparation of the first PBRER is comparable to preparing the first DSUR.
Each section of the report should be reviewed and updated. Some parts of the report might remain the same but usually there are several sections where data is updated with the latest information.
Benefit-Risk Assessment
Benefit-risk assessment consists of reviewing the new information from the interval and updated cumulative data, assessing the new data sets and writing descriptions of the assessments.
Benefit-risk assessment for a periodic safety report should be performed by a pharmacovigilance specialist or a group of specialists who understand pharmacovigilance but also the product and indication from a medical perspective.
Benefit-risk assessment is important for establishing baseline efficacy and effectiveness information. The information for the baseline comes from literature review, pre-clinical and clinical study data. This baseline data is then used for the RMP but also for the product information such as the PIL, SmPC, marketing materials etc.
Regional Implementation
Although the same template and format can be used for different regions, the data within the aggregate report might not be the same even if the same product or products are covered by the reports. This is because the data might be pointing to different things regionally.
There are regional differences in product information which means that the company needs to establish one “true source” of benefit-risk profile. The true benefit-risk profile is described in the reference product information. Reference product information is often called Company Core Data Sheet (CCDS). CCDS is used for the PBRER.
How to submit a periodic safety report?
Aggregate reports are usually a single pdf file with the table of contents, bookmarks, and hyperlinks.
According to the ICH’s electronic Common Technical Document (eCTD) standard, the periodic safety report PDF file is placed in the correct node (folder).
eCTD publishing is performed following the authority guidelines:
- gateway connection
- zip file containing folders
- xml file with a description of the folder structure.
For authorized medicinal products, the eCTD format is mandatory to use for all regulatory submissions within all procedure types within EU/EEA and it is the main way of submission in the US as well.
DSUR submission
Following the EU Clinical Trials Regulation, the submission of DSURs is performed in CTIS for trials conducted in EU countries. CTIS is an online portal for clinical trial sponsors and other organizations to apply to carry out a trial in the EEA, submit data related to a clinical trial and post trial results. To learn more about CTIS, check out our blog post about the topic and euclinicaltrials.eu.
In the Annual safety reporting section of the Sponsor workspace of CTIS, the user can click the “New ASR” button and enter information about the organization and the trial. Then the user adds the information about the annual safety report (DSUR) and attaches the DSUR file.
In the EU, the new CTIS system can be used to submit the DSUR. This is a simple process where a dedicated form is filled in with administrative information and the prepared DSUR pdf file is uploaded to the system.
In the US, the DSUR is submitted to the FDA using eCTD publishing and Gateway submission through the FDA Electronic Submissions Gateway (ESG).
PSUR submission
Following the EU Clinical Trials Regulation, the submission of DSURs is performed in CTIS for trials conducted in EU countries. CTIS is an online portal for clinical trial sponsors and other organizations to apply to carry out a trial in the EEA, submit data related to a clinical trial and post trial results. To learn more about CTIS, check out our blog post about the topic and euclinicaltrials.eu.
In the Annual safety reporting section of the Sponsor workspace of CTIS, the user can click the “New ASR” button and enter information about the organization and the trial. Then the user adds the information about the annual safety report (DSUR) and attaches the DSUR file.
In the EU, the new CTIS system can be used to submit the DSUR. This is a simple process where a dedicated form is filled in with administrative information and the prepared DSUR pdf file is uploaded to the system.
In the US, the DSUR is submitted to the FDA using eCTD publishing and Gateway submission through the FDA Electronic Submissions Gateway (ESG).
PSUR Submission Requirements in the EU
Marketing authorization holders are legally required to submit PSURs, in line with Article 107b of Directive 2001/83/EC, Regulation (EU) No 1235/2010, Directive 2010/84/EU and Commission Implementing Regulation (EU) No 520/2012.
eCTD instructions for PSUR
The requirement is that all PSURs in the EU must be submitted to the central PSUR repository using the eSubmission Gateway / Web Client. Use of the PSUR repository is mandatory for both centrally and nationally authorized medicines, whether they follow the EU single assessment or a purely national assessment procedure.
While this might sound like there’s a website or a system similar to CTIS where you can submit the PSUR pdf file, in fact, this is not the case. The PSUR eSubmission portal is only used to input the PSUR process and company related administrative information and to generate a delivery file (XML file containing the word “delivery”). This file is then added to the top-level (root) folder in the document package (ZIP file).
Administrative information for PSUR
Marketing authorization holders are required to fill in all the submission attributes through the eSubmission delivery file UI (eSubmission Gateway). This delivery file should be completed in accordance with the published EURD list, where the procedure number is the combination of a unique ID and the applicable Data Lock Point (DLP) in YYYYMM format.
The use of the XML delivery file for submissions to the PSUR Repository is mandatory for all PSURs and any related eCTD sequence submissions via the eSubmission Gateway and/or the Web Client.
Submission using the eSubmission Gateway Web Client
EMA’s eSubmission Gateway Web Client is (formerly Axway’s, nowadays independent company) Synplicity software which is essentially a cloud platform where the user registers through EMA’s registration system. After that, the submissions work pretty much the same way as with any cloud based file sharing with a built submission automation that sends the files using a gateway solution.
- First time users need to perform registration at
https://esubregistration.ema.europa.eu/registration/ - Reset password at https://eu.syncplicity.com
- Access the eSubmission Gateway Webclient
- Add the zip folder to the outbox folder which triggers the submission
- The file is sent through the gateway from the outbox folder, and moved to the archive folder
- When the submission is received by the gateway, an MDN text file is uploaded to the inbox folder
- When the submission package passes EMA’s review system, an acknowledgement file is uploaded to the inbox folder
For more detailed information about the usage of the eSubmission Gateway Web Client, see the instructions here.
Frequency of Periodic Safety Reporting
It’s normal that after a new product has been approved aggregate safety reports are required frequently.
Once the product has been on the market for a longer time, for example 3 or 5 years, and no new risks have been identified during this time, the frequency of the periodic reports is decreased to every 3 years, for example.
For generic and biosimilar products the frequency can be lower from beginning if the originator product has been on the market for a longer time. In some situations there are no regular aggregate reports required if the originator product has been on the market for decades, for example paracetamol and ibuprofen products.
Depending on the markets, where the product is authorized the frequency of the aggregate reports may vary. Often the countries, especially ICH members, have tried to harmonize the timing of the reporting and also with few adjustments the same core information in the PSUR can be used to create market specific reports.
DSUR Frequency
The rule of thumb for clinical trial period is that an annual safety report is always required, whether it be DSUR or other format.
PSUR Frequency in EU
Article 107c of EU Directive 2001/83 states that the marketing authorization holder must submit PSUR when requested by the national competent authority or following schedule:
- If the product has not yet been placed on the market, at least every 6 months following authorization and until the placing on the market
- If the product has been placed on the market, at least every 6 months during the first 2 years following the initial placing on the market
- Once a year for the following 2 years
- At three-yearly intervals thereafter
A different frequency may be assigned as a condition of the authorization.
Generic products, well established use products, homeopathics and traditional herbal medicines are excluded from the legal requirement, unless there is a condition in the marketing authorization for PSURs to be submitted or PSUR submissions are requested by EU Member States on grounds of pharmacovigilance.
European Union reference dates (EURD) list
Marketing Authorization Holders (MAH) for active substances and combinations of active substances that are subject to assessment at EU level must submit the PSURs following the reference dates (EURD) list. The list is published as an Excel file on the EMA website.
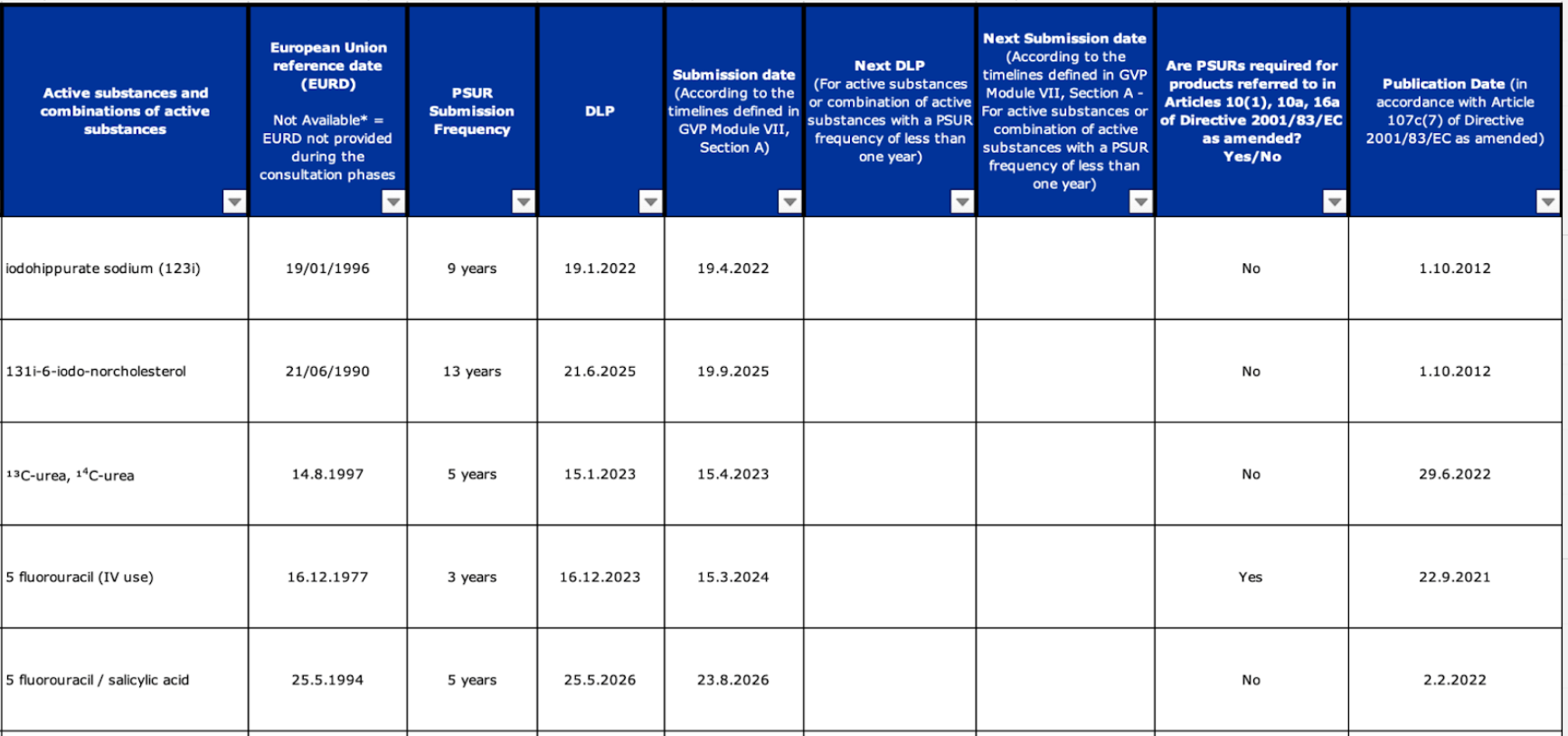
EURD list
The EURD list is a legally binding document and MAHs are legally responsible for complying with its requirements. The list overrules the “standard” PSUR submission cycle and any conditions related to the frequency of PSUR submission included in a marketing authorization.
According to Article 107c (7) of the Directive 2001/83/EC six months are needed from the EURD list publication date for the PSUR submission date to become legally binding.
In those cases where the first submission date of a newly nationally authorized product falls within the first six months from the date of the relevant EURD list publication the EURD list will reflect the second data lock point (DLP) and submission date. The first PSUR has to be assessed at a national level. EU Member States have agreed to review this first PSUR in an informal work sharing procedure.
PSUR Frequency for nationally assessed
substances in the EU
The EURD list does not include substances assessed at national level in the EU. For active substances for which the submission of the PSUR is done nationally, the frequency of submission is determined at national level by the NCA.
Frequency of postmarketing periodic reporting
in the United States
Regarding PADER in the US, the Title 21 CFR Chapter I Subchapter D Part 314 subpart B Section 314.80 on Postmarketing reporting of adverse drug experiences, says that the applicant must report each adverse drug experience not serious and unexpected at quarterly intervals, for 3 years from the date of approval of the application, and then at annual intervals. For clarity, it should be mentioned that also serious unexpected adverse drug reactions are included in the PADER.
The same legislation states that the applicant must submit each quarterly report within 30 days of the close of the quarter (the first quarter beginning on the date of approval of the application) and each annual report within 60 days of the anniversary date of approval of the application. Upon written notice, FDA may extend or reestablish the requirement that an applicant submit quarterly reports, or require that the applicant submit reports under this section at different times than those stated.
PSUR single assessment procedures (PSUSAs)
The Pharmacovigilance Risk Assessment Committee (PRAC) is in charge of issuing recommendation on the PSUR assessment for a single centrally authorized product and of the EU PSUR single assessment process.
The purpose of the EURD list is to harmonize data lock points and frequency of submission of PSURs for medicines containing the same active substances or combinations of active substances. Submission of the PSURs simultaneously allows the single assessment of related PSURs to take place at a European level.
EMA and national competent authorities (NCA) assess information in PSURs to determine if there are new risks identified for a medicine and if its risk-benefit balance has changed. A PSUR assessment can determine if further investigations on a specific issue are needed, or if an action is necessary to protect public health. This could be for example an update of the information provided to healthcare professionals (risk minimization materials) and patients (product information leaflet).
Conclusion
It should be clear that periodic safety reporting is an important, if not the most important part of pharmacovigilance. The purpose is for the study sponsors and marketing authorization holders to regularly assess and report the risk-benefit profile to the regulators so that they can confirm that the study can continue or that the drug is safe enough to keep on the market.
Despite the various regulations globally, the periodicity and format of the aggregate reports are quite well harmonized thanks to the guidelines from ICH and the EURD list from EMA that is recognized in other markets as well.
There are still some challenges related to the periodic reports. For example, the submission process varies based on the territory. There are also quite a lot of different special terms related to the aggregate reports, and local variations.
Probably it does not come as a surprise for those who have read Tepsivo’s other blog posts that we have several ideas how this process could be improved. One thing is to use Tepsivo Platform to centralize the safety information, product information and other information related to the PV system for easy generation of the main structure of the DSUR and PBRER and to use Tepsivo Safety Database for quickly creating all the necessary aggregate tables for the appendices.
If we think about the whole process of the periodic reporting, there is a lot of room for improvement. There is a lot of duplicity with marketing authorization holders with the same active ingredients reporting similar or exactly the same information. Similar or exactly the same periodic reports are assessed by several different authorities around the world. The current technology already in use in most of the pharmaceutical organizations would (and does) allow continuous updates of the full safety information and risk-benefit monitoring to a central location. In this central location (think of WHO VigiBase) data could be collected by several different MAHs and accessed centrally by one authority’s representative assigned to that product or product group. These assessments could be then approved globally.
Did you like the article? Share with your network!
…or tell us your opinion.

Follow our newsletter!
Keep up with industry trends and get interesting reads like this one 1x per month into your inbox.
Learn more about Tepsivo
We deliver modern PV solutions to fulfill your regulatory needs using less resources. See how we do it >

0 Comments