No time to read now?
-> Download the article as a handy pdf
List of contents
What is IDMP?
A comprehensive summary
Martti Ahtola | Aug 18, 2022

What is IDMP?
IDMP stands for Identification of Medicinal Product standard which describes the concept of identifiers, medicinal product terms, and their definitions. IDMP comprises of five ISO standards which describe the product information in an international framework and provide unique identifications of medicinal products with consistent documentation and terminologies. If you are new to the subject, this still doesn’t give a full explanation why it seems that everyone is talking about IDMP. Not to worry, if you read below, all will be explained. Another term that keeps popping up separately or together with IDMP is SPOR. SPOR is the acronym European Medicines Agency (EMA) has given to the implementation process of the IDMP standards. The implementation has been ongoing in a way since 2012 and it is still very much ongoing in 2022. The IDMP implementation aims to ensure clarity of exchanged information between global regulators and stakeholders. The IDMP standards support data harmonization at medicine regulatory agencies globally. The IDMP standards describe not only the content of packed medicinal product from box to drug ingredients, but they also include the registration details related to the products, for example approval date and registration number. Adverse drug reactions can be prevented or reduced after implementation of IDMP standards with the help from improved quality of data that is used in signal management. IDMP standard implementation should also speed up the international communication and help to make decisions faster at the authorities. Falsified medicines should have harder time entering the markets where IDMP standards have been implemented and the standards should prove helpful during Good Manufacturing Practice (GMP) inspections.Introduction
EMA is in the middle of implementation process for standards of the International Organisation for Standardisation (ISO) for the Identification of Medicinal Products (IDMP). The goal is to specify the use of standardized definitions for the identification and description of medicinal products for human use. The implementation and use of these standards is a regulatory requirement for the European authorities and pharmaceutical industry as they are mandated by the EU legislation. The legal requirements for IDMP implementation are described in the Commission Implementing Regulation (EU) No 520/2012 articles 25 and 26. At EMA, the implementation of the standards takes place in a phased program based on the so-called four domains of master data in pharmaceutical regulatory processes: substance, product, organization and referential (SPOR) master data.- Substance Management Service (SMS)
- Product Management Service (PMS)
- Organisation Management Service (OMS)
- Referentials Management Service (RMS)
Organisation Management Service (OMS)
The OMS part of SPOR is already functioning and it supplies organizational master data to the systems by EMA. To begin almost any regulatory process with EMA, marketing authorization holder (MAH) or applicant organization must be registered with the OMS. To manage MAH data within OMS or in the upcoming PMS, an industry user must be registered in the EMA Account Management portal (IAM) and affiliated to a specific organization with the required user role(s). OMS enables organization data to be entered once and reused many times in other business processes and related regulatory procedures.Referentials Management Service (RMS)
Like OMS, RMS is already in use, and it enables referential data to be entered once and reused many times in other business processes and related regulatory procedures. Different to OMS, the RMS data is mainly maintained by EMA, however NCAs and MAHs have the possibility to request / make changes to the referentials data. The lists of terms in RMS are used for example to provide the EDQM Standard Terms for E2B R3 ICSR forms submitted in the EU. Later, the RMS will also have a big role in storing other terms that are used to define product information in PMS. Once the updated systems are in place, RMS will supply master data to the electronic application forms (eAFs) for submitting applications for initial marketing authorizations, variations, and renewals for human and veterinary medicines. Users can select information supplied directly by these master data services when preparing their regulatory submission.Substance Management Service (SMS)
EMA has not shared much information about the functioning or the implementation of substance management service. SMS will provide a central dictionary of substance data in multiple languages. SMS is planned to support the continuous exchange of data between information systems across the European medicines regulatory network and across the pharmaceutical industry. The first version of SMS is in use by EMA and information can be submitted about substance terms through EMA Service Desk.Product Management Service (PMS)
Like the substance management service, the product management service is not yet in use. But unlike SMS, there is plenty of information available on how PMS will work and how it will be implemented (read more below). PMS will be the central database for product information, and it will make it possible to reuse the product information in different systems. Importantly, PMS makes it possible to reuse data from other parts of SPOR: OMS, RMS, and SMS, and complete the cycle.Goal of ISO IDPM Implementation
ISO IDMP standards provide a common language and share terminologies which should simplify the data sharing process between the patient, regulatory, pharmacovigilance, and other personnel of authorities and industry. In the words of the EMA, IDMP data standards provide an opportunity to link the regulatory world with the supply chain world through linkage of the systems and re-usage of data.
The purpose of these standards is to facilitate the reliable exchange of medicinal product information in a robust and consistent manner, by providing the common product ’language’ for stakeholders to use in their interactions. The authorities have stated that standardized data alone is not sufficient to achieve these benefits. The benefits of IDMP would be realized incrementally as all phases of SPOR are completed and if other opportunities for integration are implemented.
Currently, there are many different dictionaries in use by different authorities (EMA, WHO, National Competent Authorities (NCA)) and the industry.

UNICOM’s interpretation of the Flow of ICSRs and Drug Dictionaries used
Benefits of IDMP
In addition to general simplification, using ISO IDMP within regulatory activities is expected to bring also other benefits to regulators, industry, and, ultimately, the patients. Using high-level terminology, the authorities have imagined that this will happen by:- Facilitating the identification and exchange of product and substance information globally, across regulators
- Improving data integrity and reliability
- Enabling reuse of data across different procedures and regulators
- Reducing silos and improving interoperability across EU systems through optimization and simplification of data operating models and data management practices
- Streamlining, optimizing, and simplifying regulatory processes to fulfil regulatory requirements more efficiently
- Speeding up decision-making and improving communication with the stakeholders through easily accessible and highly reliable data.
IDMP Throughout the product Lifecycle
ISO IDMP is planned to cover the products all the way from the manufacturing site to patients’ hands, but it also covers the entire product lifecycle:
- products in development
- investigational products
- products under evaluation
- authorized products.
- Streamlining, optimizing, and simplifying regulatory processes to fulfil regulatory requirements more efficiently
ISO IDMP has multiple use cases within the regulatory context. Some examples provided by the authorities are listed here:
Pharmacovigilance
Having harmonized product definitions supports signal detection activities by improving the quality of data used for signal management. Standardized terminology would also speed up communication, decision-making, and actions.
Regulatory submissions
Allowing consistent information on medicinal products to be shared and re-used across different procedures and among various regulators would reduce the duplication of efforts that is currently required in regulatory affairs work.
Clinical trials
Stakeholders can access clinical trial data using agreed and well-supported standards which should improve the assessment and scientific evaluation of the product.
Good Manufacturing Practices (GMP) inspections
Inspections on manufacturing sites are based on accessible information. IDMP standards are imagined streamlining GMP inspections, particularly for urgent situations involving defects. The standards should also help with detecting falsified products and preventing their entry to the delivery chain.
Legislative Background
The legal requirements for the implementation of these standards are described in Commission Implementing Regulation (EU) No 520/2012 articles 25 and 26. Article 25 describes the use of internationally agreed terminology for the classification, retrieval, presentation, risk-benefit evaluation and assessment, electronic exchange and communication of pharmacovigilance and medicinal product information. In addition to IDMP ISO standards, this article defines the requirements for the use of MedDRA and the list of Standard Terms published by the European Pharmacopoeia Commission (EDQM). Article 26 describes the use of internationally agreed formats and standards for the classification, retrieval, presentation, risk-benefit evaluation and assessment, electronic exchange and communication of pharmacovigilance and medicinal product information. In addition to the possible (may apply) implementation of the IDMP standards, the article defines the requirements for the use of the Extended EudraVigilance Medicinal Product Report Message (XEVPRM), ICH E2B(R2) and ICH M2 standard Electronic Transmission of Individual Case Safety Reports Message Specification (eCTD).ISO IDMP standards
So, what do the standards actually contain? According to EMA’s introduction to ISO IDMP and SPOR program, the ISO IDMP standards establish definitions and concepts and describe data elements and their structural relationships.
IDMP is composed of five separate ISO standards. The five standards cover the following aspects to describe a medicinal product:
- Medicinal product name
- Ingredient substances
- Pharmaceutical product (route of administration, strength)
- Marketing Authorization
- Clinical particulars
- Packaging
- Manufacturing
The standards are:
- Substance Identification (SubID) ISO 11238: Data elements and structures for unique identification and exchange of regulated information on Substances
- Dosage Form and Route of Administration ISO 11239: Data elements and structures for unique identification and exchange of regulated information on pharmaceutical dose forms, units of presentation, routes of administration and packaging (EDQM data standards)
-> Already in use for ICH E2B R3 since 1st of July 2022 - Units of Measurement ISO 11240: Data elements and structures for unique identification and exchange of units of measurement (UCUM)
-> Already in use for ICH E2B R3 - Pharmaceutical Product Identifier (PhPID) ISO 11616: Data elements and structures for unique identification and exchange of regulated pharmaceutical Product information
- Medicinal Product Identification (MPID) ISO 11615: Data elements and structures for unique identification and exchange of regulated medicinal Product information. ISO IDMP standards apply to Human medicinal products and investigational medicinal products
Specifications – Technical Implementation of ISO IDMP
In addition to the ISO IDMP Standards, additional, more detailed specifications and guidance are provided to implement ISO IDMP successfully at an organization.
- ISO IDMP Implementation Guides (Technical Specifications) that provide business-level description of IDMP: define the technical details on how to implement the standards, such as specific fields, their formats, and business rules describing their use.
- HL7 messaging specifications: define the messages that will be used to exchange IDMP information, which are based on HL7 (Health Level Seven) standards.
- EU Implementation Guide (EU IG): provides guidance on the interpretation of data fields specifically for the EU regulatory environment as well as guidance on the processes for submitting and updating data.
ISO IDMP Implementation Guides
In addition to the five ISO standards, EMA uses in their SPOR process the following ISO technical specifications that provide implementation guidelines for the standards:
- ISO/TS 19844: Implementation guidelines for data elements and structures for the unique identification and exchange of regulated information on substances
- ISO/TS 20440: Implementation guide for ISO 11239 data elements and structures for the unique identification and exchange of regulated information on pharmaceutical dose forms, units of presentation, routes of administration and packaging-> Already in use for ICH E2B R3 since 1st of July 2022
- ISO/TS 20443: Implementation guide for ISO 11615 data elements and structures for the unique identification and exchange of regulated Medicinal Product information
- ISO/TS 20451: Implementation guide for ISO 11616 data elements and structures for the unique identification and exchange of regulated pharmaceutical product information
Specification for Information Exchange
The draft standard for international messaging known as Fast Healthcare Interoperability Resources (FHIR, which apparently is pronounced as “fire”) is being used as the basis for the application programming interface (API) for the SPOR APIs.
SPOR RMS API is already in use, and it has been implemented by the industry for EDQM Standard Terms (SPOR RMS API) effective from July 1, 2022. Similar FHIR-based API is planned for PMS (and presumably for SMS, however, this is not mentioned in the EMA documents).
FHIR is the data standard that supports the exchange of information about medicinal products, substances, and related referential data in the European medicines regulatory network.
EMA, the United States Food and Drug Administration (FDA) and the European medicines regulatory network are working with HL7 to incorporate the ISO IDMP standards into the FHIR specification.
EU IDMP Implementation Guide
EU IDMP Implementation Guide (EU IG) for the submission of data on medicinal products defines the implementation requirements of the ISO IDMP standards for the European Union. This implementation guide is the basis for submitting and exchanging medicinal product data in the EU. Its purpose is to enable stakeholders to prepare for the implementation of ISO IDMP standards.
The EU IG has been prepared by EMA after consultation with different stakeholders: representatives of MAHs and sponsors, NCAs, industry associations, international public organizations and software vendors through the SPOR Task Force and the EU Telematics governance.
EU IG provides information on the following:
- Timelines
- Requirements
- Process
- Technical specification
- Data elements
- Associated business rules
The EU Implementation Guide is composed of the chapters below:
- Introduction – EU Implementation Guide
- Chapter 1 – Registration requirements
- Chapter 2 – Data elements for the electronic submission of information on medicinal products for human use
- Chapter 3 – Process for the electronic submission of medicinal product information (not available)
- Chapter 4 – Data quality assurance (not available)
- Chapter 5 – Data access/export (not available)
- Chapter 6 – Technical specifications on structure and format: Technical specifications for the API, contains description of principles, security, resources, calls, end-points
- Chapter 7 – Migration guide: migration rules between xEVMPD and PMS including backwards compatibility rules
- Chapter 8 – Practical examples
- Chapter 9 – Process for submitting existing data on medicinal products authorised for human use (not available)
Updates to the EU Implementation Guide
The European medicines regulatory network has agreed on a phased release plan for the EU Implementation Guide.
EMA hopes to have a close collaboration between all stakeholders in the preparation activities, including industry, software vendors and regulators. That sounds good, however, seeing that Tepsivo has been proactive on the topic, and we have not found any possibilities for interaction as a software vendor and MAH representative, it may be best to assume that that ship has sailed.
EMA published the first version of the EU IDMP Implementation Guide (‘EU IG v1’) in February 2020. It provided early information to help stakeholders plan and prepare their implementation of ISO IDMP standards in the EU.
EMA published the second version (‘EU IG v2.0’) in February 2021. The EU IG v2 supports the implementation of PMS Step 1 and the data submission of medicinal products authorized under the centralized authorization procedure (CAP).
EMA published a minor release of the guide (‘EU IG v2.1’) in June 2021 and was planning to release another minor release (‘EU IG v2.2’) later in 2021, but as of July 2022, it has not happened.
During 2022 and 2023, EMA was supposed to be working on further minor updates to version 2 of the implementation guide. However, they’re supposed to publish v2.2 before that.
To minimize the impact on implementation, EU IG v2.1 focused on enhancing data-related aspects to provide visibility of data elements, business rules, data transformation and required data collection, whereas EU IG v2.2 is supposed to focus mostly on process clarifications for the centralized procedure and enhancing the quality of the EU IG.
EU IG v2.1 contains minor updates to the data elements that should be reported to PMS, further details on the RMS lists that are needed, and more examples. EU IG v2.1 also contains new data elements that are required to support the PMS Target Operating Model foreseen in PMS Step 2. These new data elements are part of ISO IDMP standards, have optional or conditional submission, and address future integration with electronic application forms (eAF) and reporting on marketing status to support the handling of medicine shortages.
The information available now in EU IG v2.1 should remain stable. But, EMA has warned that there might be changes to field business rules, submission process, and the API specification.
EU IG v2.1 provides further detail and examples applicable to the submission process, particularly on the provenance of the different submission types.
The information in the latest EU IG enables the European medicines regulatory network to prepare for the submission of data on all medicinal products for human use authorized in the EU. The idea is that these guides offer a basis for practical preparation activities, such as:
- Performing proofs-of-concept on end-to-end processes involving the generation and submission of FHIR messages, validation, and interaction with eCTD
- Testing the use cases.
Unless you are a regulatory authority or very deeply involved in the preparation of this initiative (for example belonged to the above-mentioned task force), which we assume that only a handful of the biggest players in the industry are, these expectations for companies to start preparing based on the currently shared information may seem far-fetched.
EMA will publish more chapters as part of the guide’s next major release, EU IG v3, to help the European medicines regulatory network prepare for Step 2; the last step of product data implementation.
Phased implementation of the SPOR program
Pharmaceutical companies will be required to submit data to EMA in accordance with the ISO IDMP standards, following a phased implementation of the SPOR program. The implementation of SPOR requires a coordinated program involving all key stakeholders, including EMA, national competent authorities, industry representatives and software vendors. The European Commission, European Union Network Data Board and EU ISO IDMP Task Force have endorsed a phased implementation of the ISO IDMP standards. This will allow lessons learnt during each phase to be applied to subsequent phases, processes, and systems to mature over time and stakeholders to gain an understanding prior to the full roll out. The submission and maintenance of data on authorized human medicines is already mandatory since July 2012. This process is based on a format called Extended EudraVigilance Product Report Message (XEVPRM), A.K.A. Article 57 format and related database (XEVMPD), which will be replaced by the ISO IDMP compatible format when the implementation process is complete. This SPOR transition has been ongoing since 2016. The first phase of SPOR implementation focused on delivering the RMS and OMS, which lay the data foundations for the subsequent delivery of PMS and SMS.IDMP Implementation Timeline
The EMA will be the first health agency to mandate compliance with ISO IDMP. In some of the official communication it has been said that the FDA is not far behind in the implementation. However, the E2B R3 adoption seems to be delayed which would indicate delays in other parts of the information structure updates. FDA’s own information site on ISO IDMP only mentions that FDA is involved in the international collaboration.
In theory, the publication of the EU Implementation Guide v2 in February 2021 started a 2-year implementation window of IDMP. But, it seems that this implementation window has been extended to unknown length.
Originally, it was expected that in Q1 2022, Pharma companies would be able to submit product data in IDMP format, and by Q1 2023, this would become mandatory.
Yet, in Q3 of 2022 there is no information about the possibility to submit product information in IDMP format. So, we would guess that with the current implementation pace, which is unlikely to change, the new format would become mandatory maybe late 2024 or 2025 earliest.
While we have our own guestimates, according to the latest information EMA expects to progress the following activities in 2022:
- Make ISO IDMP-compatible product data available on all authorized medicinal products in the EU, including both centrally authorized products and non-centrally authorized products.
-> This will result from a data migration and continuous updates from the EMA database (SIAMED) and the xEVMPD (Article 57) database to the PMS following the ISO IDMP standards - Enable pharmaceutical companies to correct and complete PMS product data
- Enable data approved within a regulatory application to be stored in the PMS
- Ensure adequate data quality in the PMS so that it can be confidently reused across procedures.
While the wheel is slowly turning, EMA expects the marketing authorization holders and national competent authorities to:
- Align their systems with the released terminologies for referentials, organisation and substance data
- Request the registration of any new or missing controlled vocabulary required for the submission of medicinal product data
- Start structuring their product data according to the rules described in chapter 2 of the EU Implementation Guide.
It’s not surprising that things at EMA, being a regulatory body, tend to move a little slow. We all know how quick regulatory agencies are; still keeping Brexit in fresh memory, as an example.
Updates to eAF – DADI
EMA’s electronic application forms (eAF) facilitate capturing data for regulatory processes. EMA is updating the eAFs to improve the process of entering product data with the DADI project. In 2022, EMA is focusing on replacing the current pdf-based electronic applications forms by web-based forms. The first form to be replaced is the variation eAF. Forms supporting other regulatory procedures, such as the initial marketing authorization, will follow.
An updated implementation timeline and call for volunteers for user acceptance testing for DADI were recently published. At the moment, it is expected that the transition period for new human variations eAF will start in October 2022.
EMA has suggested that marketing authorization applicants can start applying the product management rules described in the implementation guide to structure their product data. EMA is suggesting that this would enable the marketing authorization applicants to complete and correct any product data as necessary before the use of the DADI web forms becomes mandatory at the end of a transition period.
However, it is very important to note that the eAF transition period planned to end in March 2023 applies only to variations, not marketing authorization applications. The development of the web form for initial marketing authorizations is planned to start during the Q4 2022 and last at least until end of 2023. Meaning that the application form would be available maybe in 2024. Marketing authorization holders should not rush to update their product information management processes during this year, most likely not even the next.
OMS Implementation
Organisation Management Service (OMS) has already been implemented and it is used for several EMA systems, for example EudraVigilance and SPOR RMS API. Updates to the system will be necessary, although, it is unlikely that these would affect the stakeholders.
RMS Implementation
RMS is the second part of SPOR that has been already implemented and is available for use. The API connection to RMS is used to retrieve EDQM standard term dictionaries and it can be assumed that similar implementation can be used to implement the use of the other dictionaries in RMS.
SMS Implementation
Product Management Service (PMS) and Substance Management Service (SMS) are the two parts of SPOR that will heavily support regulatory activities in the EU. According to the current timeline, the implementation of SMS should be finalized by the end of 2024.
The PMS and SMS implementation process is iterative. The first iteration covers a subset of ISO IDMP data fields. Later iterations will see the standards fully implemented in the EU.
The first iteration of the SMS in 2019 enabled users to request the registration of a new substance term or the update of an existing substance term through EMA Service Desk. This allows EMA to manage the substance data. Future iterations of SMS will include synchronizing SMS with the European substance reference system (EU-SRS) database and delivering an SMS user interface.
UNICOM announced that from 12th of April 2022 onward, EMA is publishing the non-confidential substances data from SMS. Each substance will have a flag to indicate whether or not this substance was cleansed. The data is published daily in CSV format: one table containing all substances.
SMS is currently live as an EMA internal system. Until it is onboarded in the SPOR portal, external users should use the following systems:
- View and search substance data in EUTCT or IRIS
- Submit substance change requests in the EMA Service Desk portal
- Export of substance data
Currently, the information is downloadable only as a 200,000 row CSV file.
PMS Implementation
EMA and the European medicines regulatory network have agreed to a phased implementation of PMS:
- Product data preparatory phase
- PMS implementation – step 1
- PMS implementation – step 2
The product data preparatory phase focuses on getting the target operating model (TOM) ready. This phase began with the launch of the RMS and the OMS in 2017 and continues throughout 2022.
EMA is still deciding on some aspects of the PMS implementation. These undecided aspects include:
- Whether it is still relevant to follow a two-step approach (CAP & FHIR message first; non-CAPs second) and how this impacts the process described in the EU IG chapter 3.
- Whether replacing the XEVPRM format, with the new ISO IDMP compatible format place only after all relevant application forms are released by the DADI project.
EMA recommends that applicants monitor the EMA website as well as the eSubmission website for updates and announcements.
The first iteration of the PMS is planned to cover a subset of the authorized medicinal product part of the ISO IDMP standards. As part of the first iteration relevant for pharmaceutical companies, the new ISO IDMP compatible data submission format (HL7 FHIR) replaces the XEVPRM.
Future PMS iterations should implement other product data elements of the authorized medicinal product and the investigational medicinal product part of the ISO IDMP standards.
What is UNICOM?
UNICOM project was launched to provide and improve medicines identifications accurately anywhere in the world. The aim of this project is to accelerate the implementation of the ISO IDMP standards. UNICOM is financed by EU commission research and innovation program “Horizon 2020” with an estimated budget of 21 million Euros.
The project consists of 40 partners representing all the actors of the pharmaceutical product value chain. 11 participating NCAs are concretely experimenting ISO IDMP implementation. The goal of the experiments is to build a common understanding of what needs to be done towards successfully implementing IDMP and how to learn from each other, sharing best practices.
UNICOM is focused on establishing EU substance reference system (EU-SRS). This EU-SRS is used as the basis for cross-border ePrescription system which ensures the accuracy of substance in pharmaceutical products. Currently, the ePrescription system is used in only a handful of countries including Croatia, Estonia, Finland, and Portugal.
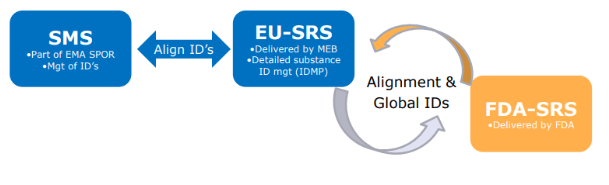
EU-SRS will be linked to SMS and to the FDA-SRS
EU-SRS was originally planned to be live by the end of 2018 and the latest due date we have found was 30th of September 2021. As far as we can tell, the system is not live yet. The implementation of EU-SRS is led by the Dutch NCA, MEB, who was recently hiring a Project Lead for EU-SRS for June 30, 2022 – January 30, 2023. UNICOM has stated that because exchange of ICSRs is highly regulated global activity, agreement at level of ICH is needed before the use of MPIDs, PhPIDs, and (S)SIDs can be started.
Aim of UNICOM
UNICOM tries to overcome the challenges in identification of medicinal product and aim to diffuse ISO IDMP standards.
- It improves pharmacovigilance detection and prevention of medicine related problems
- Delivering equivalent drugs or substitutions in cross-border services through ePrescription or eDispensation
- UNICOM also helps to integrate the real-world health data and cover the full lifecycle of a medicine.
Through this project, EU, and national SPOR databases, along with EU-SRS will be implemented to be used for cross-border ePrescription system.
IDMP Training
EMA is planning to develop an online training course comprising of series of presentations, videos, and step-by-step guides. Access to this training and testing has been promised to be free of charge similar to the current XEVMPD training and testing. Additional information on the location of the training materials as well as the aspects of the knowledge evaluation should be available in future updates of the implementation guide. Following a successful completion of the training and test, a ‘training confirmation’ will be issued by the EMA.
As currently with XEVMPD, the training will be mandatory for ‘Qualified Users’ and the training confirmation will be required from at least one Qualified User as part of the registration process before data submission can begin.
Migration from XEVMPD to PMS
EMA will migrate the data in XEVMPD to PMS (database-to-database migration) and it has been suggested that pharmaceutical companies start thinking how they will store their product information in the future and how the data will be transformed to support the new standard. EMA has provided their instructions and experience on the topic, but there are no strict rules that need to be followed for the data transformation and/or migration.
As a preparatory step, the underlying terminology in the Referentials Management Services (RMS), Organisation Management Services (OMS) and Substance Management Services (SMS) is mapped with the appropriate cross references to the XEVMPD (EV Codes). The EV Codes of the XEVMPD terminologies are loaded into a staging area where the EV Codes are transformed into the relevant RMS, OMS, SMS codes and terms. Then, the XEVMPD data is transformed into IDMP compatible format based on the PMS Logical Data Model (LDM) and according to the mapping rules when loading into PMS.
There will be an initial full data migration that includes all the versions of human authorized medicinal product including all the information relevant to marketing authorization status, country of authorization, sender of the product and status of the product. After the initial load, the data is periodically (hourly) synchronized from XEVMPD to PMS.
Import process is synchronized by identifying, extracting, and synchronizing the latest changes in XEVMPD. There will also be a feedback loop from PMS to XEVMPD, updating the latter’s information with the latest changes using XEVPRM messages. All the changes in PMS are coded with an EV code and transferred to XEVMPD regularly.
Product Information Data Model in PMS
Here we introduce some of the new or updated parts of the product information data model that will be implemented with PMS. We demonstrate how the updated system will take advantage of the dictionaries / lists and information stored elsewhere. Please note that the links to SPOR RMS require a SPOR registration, available now to all stakeholders.
We do not describe here all the product information fields, nor do we describe all the information available about the fields listed here. We only describe a few example fields that are interesting in the context of ISO IDMP implementation.
For the latest information about the data fields and product information data model implementation, see Product Management Service (PMS) – Implementation of International Organization for Standardization (ISO) standards for the identification of medicinal products (IDMP) in Europe, Chapter 2: Data elements for the electronic submission of information on medicinal products for human use.
The product information is categorized in the following way (this will be familiar to those who know their way around XEVMPD):
- Medicinal product
- Marketing authorization information
- Therapeutic (product) indication
- Packaged medicinal product
- Ingredients
- Pharmaceutical product
Product Management Service Identifier (PMS ID)
PMS ID will be assigned to each medicinal product single entry following the first submission of the authorized medicinal product data to PMS. It is a unique, stable, and permanent ID supplementary to any existing authorization number or equivalent identifier as assigned by an authorizing body. The PMS ID is composed only of digits.
This attribute is automatically generated by the PMS system and remains unchanged through the lifecycle of the medicinal product. The PMS ID shall be specified when performing any maintenance related activity of the medicinal product data.
Medicinal Product Identifier (MPID)
MPID will be assigned to each authorized medicinal product. It is also a supplementary ID to any existing authorization number or equivalent identifier as assigned by an authorizing body in a region. The MPID is defined by the following segments:
- Country code segment (ISO 3166-1 alpha-2 code elements)
- Marketing authorization holder (i.e. location ID) code segment
- Medicinal product code segment (i.e. unique medicinal product ID).
- Any change of the values related to these three code segments would result in the assignment of a new MPID.
- MPID is automatically generated and maintained by the PMS system.
- Pharmaceutical Form and Combined pharmaceutical dose form
The authorized pharmaceutical dose form(s) or the dose form submit is provided as an RMS term ID from the EDQM standard terms.
The combined pharmaceutical dose form(s) are also provided as an RMS term ID. Values for pharmaceutical forms and combined pharmaceutical dose forms are available in one of the SPOR RMS lists and SPOR RMS Combined pharmaceutical dose form list.
Legal status of supply
The legal status of supply is listed in the SPOR RMS Legal Status for the Supply list. Legal status of supply RMS list is to be expanded to account for the different legal status definitions at national level.
Product classification
The medicinal product can be classified according to various classification systems. In the context of PMS iteration 1, the product classification describes the following product information:
- XEVMPD Medicinal product types – not yet available
- Legal basis – SPOR RMS Application Legal Basis list
- ATC Code(s) – SPOR RMS Anatomical Therapeutic Chemical Classification System – Human list
- Medicinal product category – not yet available
PSMF
As initial temporary process, registration of the PSMF location will need the use of two systems XEVMPD and PMS. It will be performed in two steps:
1. At the time of the marketing authorization application, the applicant should submit electronically the PSMF location information using the agreed format as referred to in chapter IV, Article 26 of the Commission Implementing Regulation (EU) No 520/2012 using the XEVMPD and XEVPRM format.
a. This will generate a PSMF location reference number/identifier, which is the unique code assigned by the EMA to the master file.
2. Subsequently, the PSMF location reference number/identifier generated in XEVMPD database should be linked to the medicinal product during the submission of product data in PMS.
This process is planned to be revised in future versions of the EU implementation guide for PMS to make use of a central repository for Master File information using SPOR capabilities.
QPPV
After completion of the EU QPPV registration, the QPPV code from EudraVigilance can be referenced in PMS. The details of the QPPV are pre-registered externally to PMS, so it is not necessary to provide full details of the QPPV again in the submission of the product.
The value “Qualified Person in the EEA for Pharmacovigilance” shall be selected from the SPOR RMS Contact Party Role list.
Attached document
In case of submission of medicinal products dataset via API (e.g., NAPs or CAPs in which the FHIR message is not entered together with the eCTD closing sequence) a copy of the authorized SmPC document shall be provided to PMS as before with XEVMPD.
In the case of CAPs, in which the FHIR message is submitted together with the closing sequence of the regulatory activity, a link to the eCTD documentation can be provided, therefore there is no need for supplying additional documents together with the FHIR message. The link to the eCTD document should have the following format to point to the current document: 0002/m1/eu/13-pi/131-spclabelpl/ema/en/eu-combined.pdf
This process is detailed in EU IG Chapter 3 (which is currently unavailable) and will be updated for the PMS implementation Step 2.
Attached document type shall be selected from the SPOR RMS Product Information Document Type list.
Product cross-reference
A cross-reference to one or more medicinal products is to be made if the medicinal product has been authorized under the following legal basis: Generic application, Hybrid application, Similar biological application, or Informed consent application, or if the medicinal product is a Parallel Imported medicinal product.
The applicable value shall be selected from the term ID as listed in the SPOR RMS Product Cross Reference Type list and the applicable PMS ID is provided.
Manufacturing business operation
The Manufacturer shall be specified using the location identifier (LOC ID) linked to the organization as listed in the Organization Management System (OMS) following a successful registration of the organization’s details.
System Updates – HALMED Case Study
What updates the implementation of IDMP will require, depends heavily on the current system(s) of the national competent authority or marketing authorization holder.
If there already is a well-maintained database with logical data fields for products, substances, and packages, most likely the ISO IDMP implementation will not require a big update to the computerized system.
But if there is no coding system in place, it is not possible to use dictionaries, and as data about some aspects such as packages is only descriptive (text fields), a larger update and implementation or a completely new system might be required.
The Croatian NCA, HALMED, has written an article about their system update process which was recently published by UNICOM. Even though the details are specific to the Croatian authority’s own internal systems, the article describes well how to analyze the organization’s systems and data models and to determine what needs to be updated to comply with the ISO IDMP standards.
One of the first main decisions HALMED made was to choose between implementing an entirely new system (off-the-shelf or custom made) and updating their old system. HALMED chose to update the existing system NRL-PKL-PhV.
HALMED’s NRL-PKL-PhV system consists of three separate applications that share medicinal products database, document repository, referential lists, administration tools and integration services to other HALMED’s computerized systems.
The reasoning behind the decision to stick with the existing system was that the medicinal product data stored in NRL-PKL-PhV was not yet exchanged with other nation-wide eHealth systems and that several very complex processes were already supported by the system (and that they had invested years of development in the system.)
The update of the NRL-PKL-PhV system included
- reconstruction of the data model,
- the adaptation of the user interface,
- and the modification of the synchronization processes.
Data Model Analysis
HALMED analyzed the data models described in ISO IDMP standards. Most important standard for their analysis was the Medicinal Product Identification (MPID) ISO 11615:2017 for the unique identification of regulated medicinal product information.
Relying on the results of the analysis of processes utilizing medicinal products data, a detailed assessment of the current data model was performed, resulting in a thorough comparison with ISO IDMP standards to detect the gaps.
First gap analysis revealed two important refactoring areas:
1. Packaged Medicinal Product: Manufactured Item (and its composition) should be introduced into the data model, as well as Packaging Item (Container) and Device
2. Pharmaceutical Product needs some adjustments: mandatory specification of the modifiers for chemical substances; introduction of Specified substance (for proteins and biologicals) and Strength (Reference Strength as Presentation and Concentration strength were already implemented).
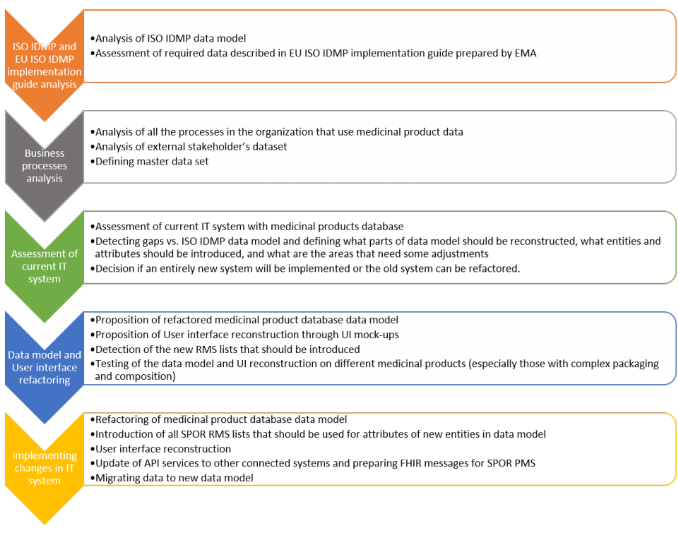
HALMED’s methodology of refactoring internal IT system and medicinal product database to comply with ISO IDMP standards
The data model and the structure of related tables enabled data entry for most entities but did not fully comply with ISO IDMP data model. Below is an example of the data model analysis for a pharmaceutical product.
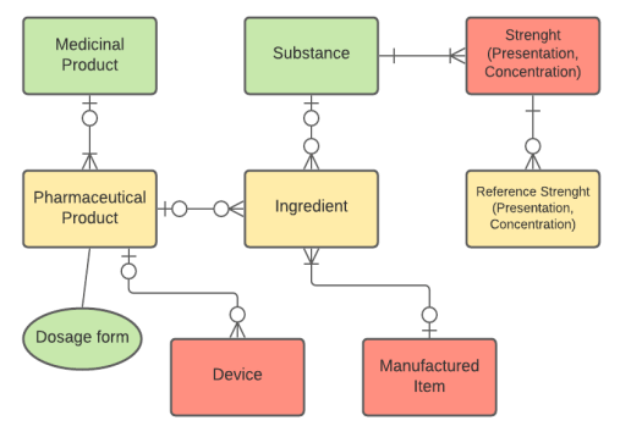
HALMED’s data model analysis of Pharmaceutical Product: Red: should be introduced in the data model, Yellow: would need adjustments, Green: already compliant with ISO standards
User Interface
As described above, user interface (UI) reconstruction was required for HALMED’s system. For example, on “Packaging” tab the following changes were required:
- Input of more Packaged Medicinal Products (i.e., outer packages)
- Input of more Package Items (Containers) with the possibility of copying data from existing Package Items
- Input of Devices with possibility of copying data from existing Devices
- Input of Manufactured Items with possibility of copying data from existing Manufactured Items
- Input of Ingredients for Manufactured Items: Substance and Specified Substance, with the Strengths and Reference Strengths (concentration and presentation)
- Copying/relating Ingredients for Manufactured Items and Ingredients for Pharmaceutical Products.
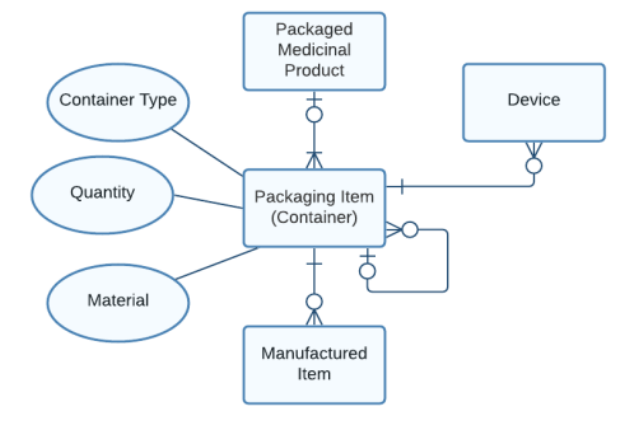
HALMED’s model for Packaging Item (Container): Containers that are referencing it as parent; can be referenced by different Devices or Manufactured items; attributes: Container Type, Material and Quantity
Dictionaries
HALMED stated that fortunately they have been using the European Union Telematics Controlled Terms (EUTCT) lists for internal codebooks from the early stages of NRL-PKL-PhV system development and those codebooks were synchronized on a daily basis. This means that the system was built to support dictionaries. Previously, it had been possible for HALMED’s users to add user-defined terms in NRL-PKL-PhV codebooks.
Planning for transition to RMS lists required assessment of different scenarios:
- Codebooks with EUTCT terms and no custom added terms
- Codebooks based on EUTCT lists and with user added terms
- Custom codebooks in NRL-PKL-PhV system
- Introduction of new RMS codebooks to NRL-PKL-PhV system (no codebooks were used previously)
Interoperability
Moreover, as part of the EU funded project eLijekovi, the future national medicinal product database in Croatia will be built in accordance with the ISO IDMP set of standards. This medicinal product database will be the foundation for all the future eHealth services related to medicinal products.
According to HALMED by complying with ISO IDMP standards, the national medicinal product database will significantly impact the Croatian eHealth system in general. It should provide an essential contribution to data interoperability throughout the Croatian healthcare systems and therefore enable a meaningful and quality data exchange between all stakeholders.
Did you like the article? Share with your network!
…or tell us your opinion.

Follow our newsletter!
Keep up with industry trends and get interesting reads like this one 1x per month into your inbox.Learn more about Tepsivo
We deliver modern PV solutions to fulfill your regulatory needs using less resources. See how we do it >

0 Comments